
The Protocol
Learn here how to extract protein-crosslinked RNA from UV-crosslinked cells in a step-to-step illustrated description of the XRNAX protocol. XRNAX extracts can be used for any of the downstream applications found in the Applications section of this website or as input for your custom protocol.
Required Material

XRNAX uses TRIZOL extraction followed by steps for solubilization, DNase digestion and isopropanol precipitation. For a maximum of 100 million cells you will need:
50 ml PBS for washing cells
8 ml TRIZOL (or TRI reagent from any other vendor)
1.6 ml chloroform
6 ml TE+SDS 0.1% (Tris-Cl 10 mM, EDTA 1 mM, SDS 0.1 %)
6 ml TE+SDS 0.5% (Tris-Cl 10 mM, EDTA 1 mM, SDS 0.5 %)
400 µl NaCl 5 M
4 µl glycoblue (or coprecipitant from any other vendor)
6 ml isopropanol 100 %
4 ml EtOH 70 %
2 µl RNasin Plus RNase inhibitor (Promega, N2611)
200 µl DNase I buffer 10 x (NEB, M0303)
100 µl DNase I (NEB, M0303L)
1 Falcon tube 50 ml
10 low-binding tubes 2 ml
For quality control you will need:
1% agarose gel stained with SYBRsafe (invitrogen, S33102)
RNase A (Thermo, EN0531)
Tris-Cl 10 mM
Proteinase K (Thermo, EO0491)
Proteinase K buffer (tris-Cl 50 mM, EDTA 10 mM, NaCl 150 mM, SDS 1 %)
RNA loading dye 2x (Thermo, R0641 or loading dye from any other vendor)
RiboRuler HR (Thermo, SM1821 or RNA ladder from any other vendor)
Xlink

Grow cells under any desired condition and UV-crosslink them on ice.
For example, when using 254 nm crosslinking, grow MCF7 cells in a 24.5 x 24.5 cm dish until confluent. When using 365 nm crosslinking, subject cells to 100 µM 4-thiouridine (4-SU) for 16 hours prior to crosslinking.
Discard the media, wash the cells with approx. 50 ml PBS, flip the dish upright and allow any residual PBS to drain on a paper towel for 1 minute.
Transfer the dish onto ice (another 24.5 x 24.5 cm dish can serve as an ice receptacle) and crosslink with 200 mJ/cm2.
Immediately harvest cells by scraping into 10 ml ice-cold PBS and transfer to a 50 ml Falcon tube. Collect residual cells into another 10 ml ice-cold PBS, combine with the rest and spin down for 5 minutes with 1000 g at 4°C. Discard the supernatant and store the pellet at -80 °C for up to 2 weeks.
TRIZOL Lysis

If starting from a frozen pellet, allow cells to thaw on ice.
Lyse up to 100 mio. cells in 8 ml TRIZOL by pipetting up and down. Make sure to dissolve clumps by pipetting against the tube walls.
All aggregates should be dissolved. If clumps remain incubate tube for some minutes on rotating wheel and continue lysis by pipetting.
Discard Aqueous Phase
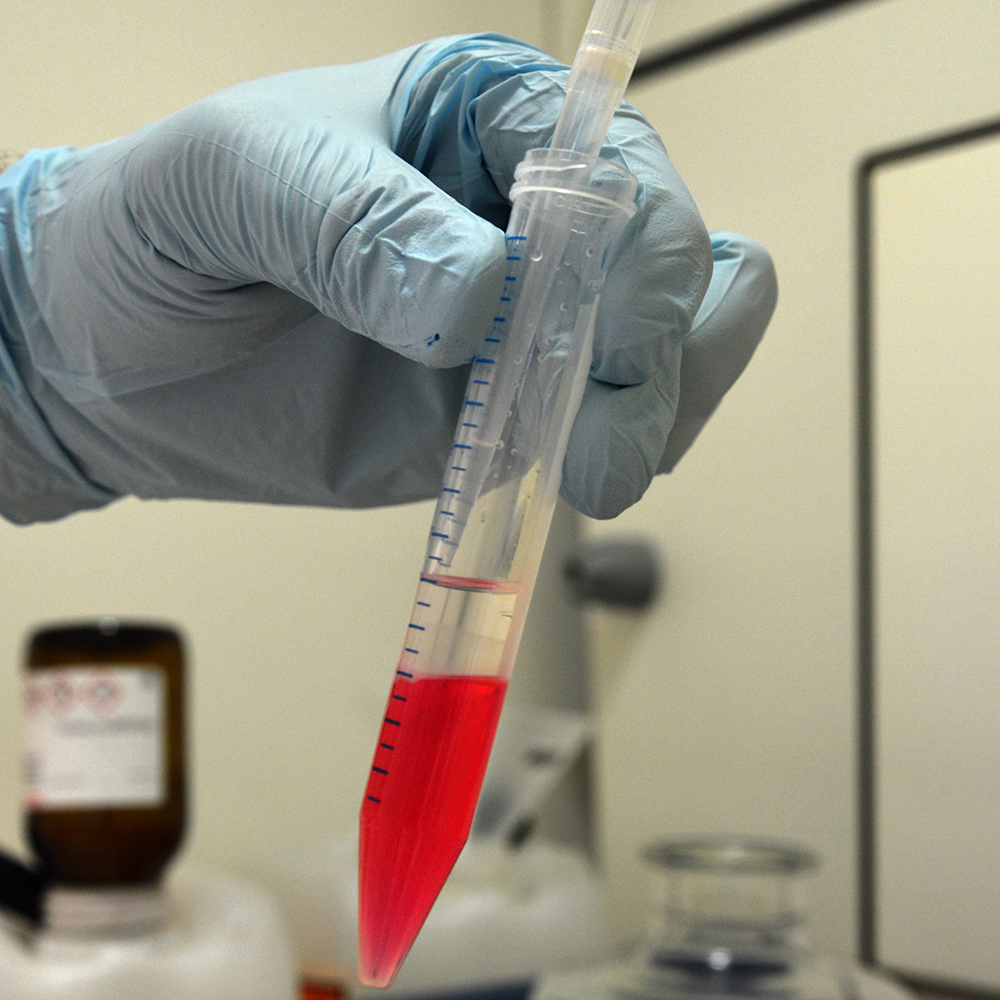
Transfer cells to a 15 ml tube and add 1.6 ml chloroform to induce phase separation. Mix by turning the tube upside down several times and let it sit for 5 minutes at room temperature.
Spin for 10 minutes with 7000 g at 4 °C.
Remove aqueous phase without disturbing the interphase. Residual aqueous phase can remain and will be washed away later.
Aspirate Interphase

The interphase of crosslinked cells is sticky and can be picked up by gently aspirating it with a 1 ml pipette tip. Transfer the interphase in one or several pieces to a fresh 2 ml tube.
Wash Interphase
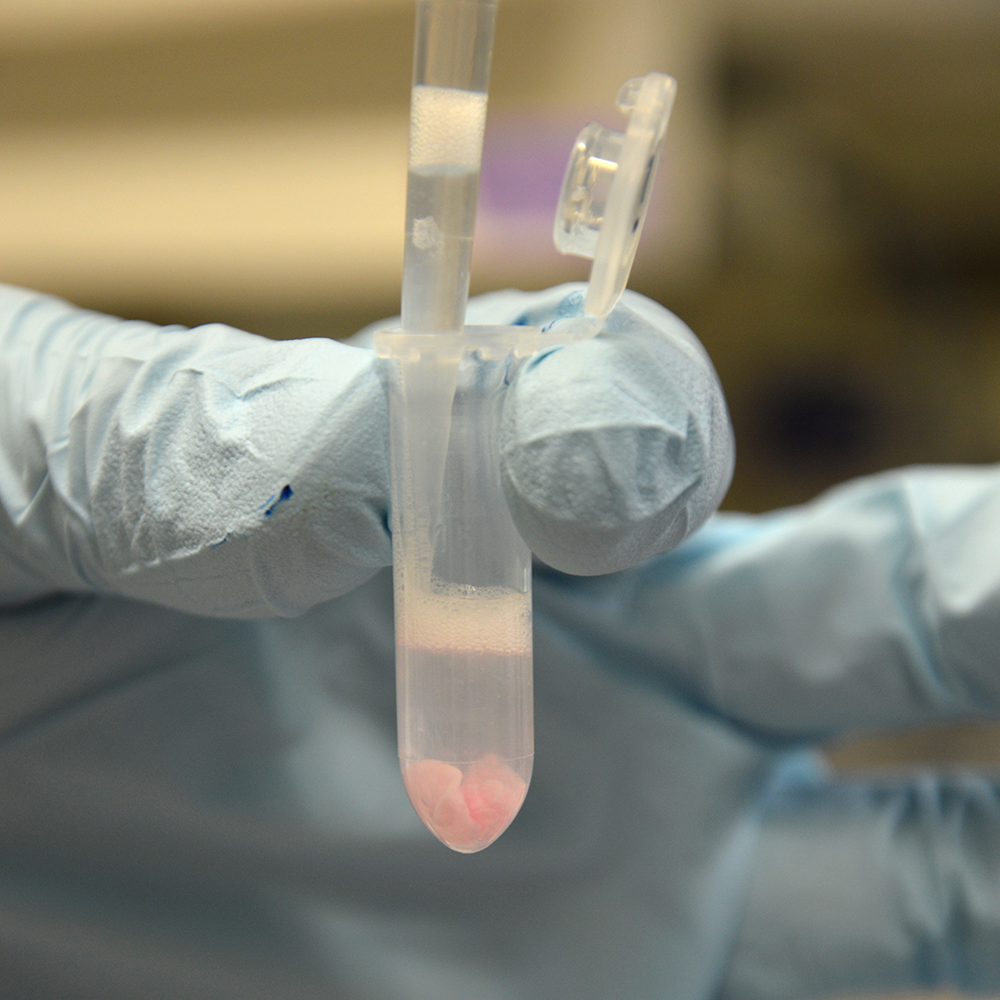
Residual aqueous phase and free protein can be washed from the interphase with TE+SDS 0.1 %. After addition of 1 ml TE+SDS 0.1 % the interphase will separate into flakes, which will remain intact if they are not sucked into the pipette tip. Wash the interphase flakes without disturbing their integrity by pipetting the wash buffer in and out.
When the wash buffer turns milky discard it and repeat with another 1 ml of TE+SDS 0.1%.
Disintegrate Interphase 1

The interphase will now be dissolved in 4 steps. Each step uses 1 ml of TE+SDS 0.1 %, which will be collected as 4 fractions. Add 1 ml of TE+SDS 0.1 % and start disintegrating the interphase flakes by pipetting them in and out. This will be difficult in the beginning but becomes easier the smaller the pieces get.
After approx. 30 times pipetting pieces will not get smaller, so they are spun down with 5000 g at room temperature for 2 minutes.
Transfer Supernatant 1

Upon disintegration the interphase will hydrate more and more, thereby gaining volume and losing its pink/ white color. After the first disintegration and spinning a transparent layer will have formed on its top. Transfer the supernatant to a fresh 2 ml tube. Leave behind the sticky interphase pellet and its viscous top layer.
Disintegrate Interphase 2-4

Repeat the disintegration using another 1 ml of TE+SDS 0.1 %. After approx. 30 times pipetting most flakes will have dissolved and the solution gains in viscosity. Spin down with 5000 g at room temperature for 2 minutes.
The 3rd and 4th repetition of this step is carried out with 1 ml of TE+SDS 0.5 % instead of 0.1 % SDS.
Transfer Supernatant 2-4

After the second or third disintegration step the interphase will have lost most of its color and forms a transparent gel instead of a visible pellet. Make sure to transfer only the solubilized supernatant leaving the gel behind.
After the 4th repetition some remnants of the gel remain not solubilized. However, by that point most of the interphase has been brought into solution so that these remnants can be discarded.
Isopropanol Precipitate 1

The four fractions contain the solubilized interphase, which will now be isopropanol precipitated. Add 60 µl NaCl 5 M, 1 µl glycoblue and 1 ml isopropanol to each fraction. Mix by turning the tube upside down several times and spin down for 15 minutes with 18000 g (or full speed) at -10 °C (or coolest setting).
Ethanol Wash 1

Discard the supernatant and combine all four pellets using 1 ml EtOH 70 %. For this purpose break each pellet up by pipetting up and down and combine it with the next one. Collect residual pellet fragments using another 1 ml of EtOH 70 % and combine everything in one tube.
Spin down with 18000 g for 1 minute at room temperature to combine all pellets into one. Discard the supernatant and remove residual EtOH as good as possible using short spins in the mini centrifuge.
Hydrate & Homogenize

The interphase pellet now contains protein-crosslinked RNA but also chromatin. DNA will be removed through DNase digestion. However, therefore the pellet needs to be brought into solution. Add 1.8 ml nuclease-free water to the pellet and vortex briefly so that the pellet detaches from the tube wall. Allow the pellet to hydrate on ice for approx. 1 hour with occasional mixing by turning the tube upside down. The pellet will expand into a milky cloud turning the solution into a gel.
Completely dissociate the solution with a 1 ml pipette by pipetting up and down until no clumps are visible. The viscosity of the solution will increase tremendously and if some of the clumps do not dissolve allow hydration to occur during some more minutes on ice.
DNase Digest

For DNA digestion add 200 µl DNase buffer 10 x and mix by pipetting. Adjusting the pH with DNase buffer will decrease the viscosity so that the solution becomes easy to pipette. Add 2 µl RNase inhibitor and 100 µl DNase, mix and incubate for 90 minutes at 37 °C, 700 rpm shaking.
Isopropanol Precipitate 2

To remove deoxynucleotides and salts the digest will now be isopropanol precipitated one more time. Split the 2 ml sample onto two 2 ml tubes and add 60 µl NaCl 5 M. Add 1 ml isoproanol, mix by inverting the tube several times and spin down with 18000 g (or full speed) at -10 °C (or coldest setting) for 15 minutes.
Ethanol Wash 2

Discard the supernatant and combine the two pellets using 1 ml EtOH 70 %. Collect residual pellet fragments using another 1 ml of EtOH 70 % and combine everything in one tube.
Spin down with 18000 g (or full speed) for 1 minute at room temperature to combine all pellets into one. Discard the supernatant and remove residual EtOH as good as possible using short spins in the mini centrifuge.
Resuspend

The pellet is now salt-free and only contains protein and RNA. Resuspend the pellet in your buffer of choice.
We usually resuspend XRNAX extracts from approx. 50 mio cells in 500 µl nuclease-free water before estimating the RNA concentration on a NanoDrop photometer using a 1:5 dilution. Note that additional adsorption by protein in the extract makes this measurement an estimate, however, in our experience this estimate lies within 90 % of the true RNA content of the sample. Note that this is the concentration, which we refer to in the 'applications' section. We usually adjust the XRNAX extract to a 1000 µg RNA/ µl stock solution in nuclease free water, which we store at -80 °C for optimal preservation.
QC

Agarose gel electrophoresis gives a good assessment of RNA integrity and DNA contamintation in XRNAX extracts. A non-treated control should exhibit a distinct shift of RNA in the gel caused by crosslinked protein. This shift should be removed by proteinase K treatment. RNA in this case should show the characteristic two-band pattern indicating intact ribosomal RNA. A smear of smaller RNA fragments indicates unwanted degradation. Remaining signal in the gel after RNase digestion indicates residual DNA in the extract. This can be caused by incomplete DNase digeston.
For proteinase K digestion add 1 µg of XRNAX extract to 10 µl proteinase K buffer 2 x and top off the volume to 20 µl. Add 1 µl proteinase K and digest for 30 minutes at 60 °C, 700 rpm shaking.
For RNase digestion top off 1 µg of XRNAX extract to 20 µl with tris-Cl 10 mM. Add 1 µl RNase A and digest for 30 minutes at 37 °C, 700 rpm shaking.
For a non-treated control top off 1 µg of XRNAX extract to 20 µl with tris-Cl 10 mM.
Combine each sample with 20 µl RNA loading dye 2 x and heat to 85 °C for 2 minutes and transfer to ice. Load 10 µl on an 1 % agarose gel with 1 x SYBRsafe. Load 3 µl RiboRuler HR on both sides and run at 2 Watts for 45 minutes.